Analyzing and visualizing cell-cell communication events#
In this tutorial, we demonstrate how HoloNet can be used to analyze and visualize cell-cell communication in spatial transcriptomics data.
HoloNet needs three inputs:
- Spatial transcriptomic data (with gene expression matrix and spatial information).
In this version of HoloNet, we support the spatial data based on
AnnDataloaded from Scanpy.
- Cell-type information.
Cell-type percentages from deconvolution methods prefer to be saved in
adata.obsm['predicted_cell_type'].Categorical cell-type labels prefer to be saved in
adata.obs['cell_type'].
- Database with pairwise ligand and receptor genes.
A pandas dataframe, must contain two columns: ‘Ligand_gene_symbol’ and ‘Receptor_gene_symbol’.
Note
The tutorial mainly follows these steps:
Load spatial transcriptomic data.
Construct multi-view communication event (CE) network among single cells (each LR pair corresponds to one view).
Visualize communication based on the multi-view network.
Other analysis methods (such as clustering LR pairs).
import HoloNet as hn
import os
import pandas as pd
import numpy as np
import scanpy as sc
import matplotlib.pyplot as plt
import torch
import warnings
warnings.filterwarnings('ignore')
hn.set_figure_params(tex_fonts=False)
sc.settings.figdir = './figures/'
Data loading#
Loading example spatial transcriptomic data#
We prepare a example breast cancer dataset for users from the 10x Genomics website.
We preprocessed the dataset, including filtering, normalization and cell-type annotation.
Users can load the example dataset via HoloNet.preprocessing.load_brca_visium_10x().
adata = hn.pp.load_brca_visium_10x()
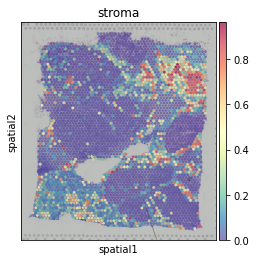
Visualize the cell-type percentages in each spot.
hn.pl.plot_cell_type_proportion(adata, plot_cell_type='stroma')
The figures outputed from HoloNet can be saved if adding fname parameter.
Note
The parameters of plotting functions in this tutorials are mainly inherited from two base plotting functions:
The cell-type label of each spot (the cell-type with maximum percentage in the spot)
sc.pl.spatial(adata, color=['cell_type'], size=1.4, alpha=0.7,
palette=hn.brca_default_color_celltype)

Loading ligand-receptor database#
We provide a database with pairwise ligand and receptor genes for users. Load the database and filter the LR pairs, requiring both ligand and receptor genes to be expressed in a certain percentage of cells (or spots).
interaction_db, cofactor_db, complex_db = hn.pp.load_lr_df(human_or_mouse='human')
expressed_lr_df = hn.pp.get_expressed_lr_df(interaction_db, complex_db, adata)
expressed_lr_df.shape
(325, 12)
We also allow access to ligand-receptor pair databases based on the MITAB canonicalized nomenclature.
We provide samples here so that users can import MITAB-compliant ligand-receptor databases and replace interaction_db
pd.set_option('display.max_columns', None)
interaction_db_MITAB = pd.read_csv('cellchatdb_human_interaction_example_with_MITAB.csv', index_col=0)
interaction_db_MITAB.head()
| interaction_name | pathway_name | ligand | receptor | agonist | antagonist | co_A_receptor | co_I_receptor | evidence | annotation | interaction_name_2 | LR_pair | #ID Interactor A | ID Interactor B | Alt IDs Interactor A | Alt IDs Interactor B | Aliases Interactor A | Aliases Interactor B | Interaction Detection Method | Publication 1st Author | Publication Identifiers | Taxid Interactor A | Taxid Interactor B | Interaction Types | Source Database | Interaction Identifiers | Confidence Values | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | TGFA_EGFR | EGF | TGFA | EGFR | NaN | NaN | NaN | NaN | KEGG: hsa04012 | Secreted Signaling | TGFA - EGFR | TGFA:EGFR | entrez gene/locuslink:1956 | entrez gene/locuslink:7039 | biogrid:108276|entrez gene/locuslink:EGFR|unip... | biogrid:112897|entrez gene/locuslink:TGFA|unip... | entrez gene/locuslink:ERBB(gene name synonym)|... | entrez gene/locuslink:TFGA(gene name synonym) | psi-mi:"MI:0018"(two hybrid) | Kotlyar M (2015) | pubmed:25402006 | taxid:9606 | taxid:9606 | psi-mi:"MI:0407"(direct interaction) | psi-mi:"MI:0463"(biogrid) | biogrid:1067440 | - |
| 1 | OSM_OSMR_IL6ST | OSM | OSM | OSMR_IL6ST | NaN | NaN | NaN | NaN | KEGG: hsa04060 | Secreted Signaling | OSM - (OSMR+IL6ST) | OSM:OSMR_IL6ST | entrez gene/locuslink:5008 | entrez gene/locuslink:3572 | biogrid:111049|entrez gene/locuslink:OSM|unipr... | biogrid:109786|entrez gene/locuslink:IL6ST|uni... | - | entrez gene/locuslink:CD130(gene name synonym)... | psi-mi:"MI:0004"(affinity chromatography techn... | Huttlin EL (2015) | pubmed:26186194 | taxid:9606 | taxid:9606 | psi-mi:"MI:0915"(physical association) | psi-mi:"MI:0463"(biogrid) | biogrid:1176790 | score:0.999999936 |
| 2 | TNFSF9_TNFRSF9 | CD137 | TNFSF9 | TNFRSF9 | NaN | NaN | NaN | NaN | KEGG: hsa04060 | Secreted Signaling | TNFSF9 - TNFRSF9 | TNFSF9:TNFRSF9 | entrez gene/locuslink:3604 | entrez gene/locuslink:8744 | biogrid:109817|entrez gene/locuslink:TNFRSF9|u... | biogrid:114281|entrez gene/locuslink:TNFSF9|un... | entrez gene/locuslink:4-1BB(gene name synonym)... | entrez gene/locuslink:4-1BB-L(gene name synony... | psi-mi:"MI:0004"(affinity chromatography techn... | Huttlin EL (2015) | pubmed:26186194 | taxid:9606 | taxid:9606 | psi-mi:"MI:0915"(physical association) | psi-mi:"MI:0463"(biogrid) | biogrid:1177155 | score:0.992349156 |
| 3 | EFNB2_EPHB2 | EPHB | EFNB2 | EPHB2 | NaN | NaN | NaN | NaN | PMID: 15114347 | Cell-Cell Contact | EFNB2 - EPHB2 | EFNB2:EPHB2 | entrez gene/locuslink:1948 | entrez gene/locuslink:2048 | biogrid:108268|entrez gene/locuslink:EFNB2|ent... | biogrid:108362|entrez gene/locuslink:EPHB2|uni... | entrez gene/locuslink:EPLG5(gene name synonym)... | entrez gene/locuslink:CAPB(gene name synonym)|... | psi-mi:"MI:0004"(affinity chromatography techn... | Huttlin EL (2015) | pubmed:26186194 | taxid:9606 | taxid:9606 | psi-mi:"MI:0915"(physical association) | psi-mi:"MI:0463"(biogrid) | biogrid:1177983 | score:0.999999507 |
| 4 | EFNB2_EPHB3 | EPHB | EFNB2 | EPHB3 | NaN | NaN | NaN | NaN | PMID: 15114347 | Cell-Cell Contact | EFNB2 - EPHB3 | EFNB2:EPHB3 | entrez gene/locuslink:1948 | entrez gene/locuslink:2049 | biogrid:108268|entrez gene/locuslink:EFNB2|ent... | biogrid:108363|entrez gene/locuslink:EPHB3|uni... | entrez gene/locuslink:EPLG5(gene name synonym)... | entrez gene/locuslink:ETK2(gene name synonym)|... | psi-mi:"MI:0004"(affinity chromatography techn... | Huttlin EL (2015) | pubmed:26186194 | taxid:9606 | taxid:9606 | psi-mi:"MI:0915"(physical association) | psi-mi:"MI:0463"(biogrid) | biogrid:1177984 | score:0.999990606 |
expressed_lr_df = hn.pp.get_expressed_lr_df(interaction_db_MITAB, complex_db, adata)
Constructing multi-view CE network#
Ligand molecules from a single source can only cover a certain region.
Before constructing multi-view communication network, we need to calculate the w_best to decide the region (‘how far is far’).
The parameters for culcalating w_best is shown in HoloNet.tools.default_w_visium().
w_best = hn.tl.default_w_visium(adata)
hn.pl.select_w(adata, w_best=w_best)

Based on w_best, we can build up the multi-view communication network.
We calculate the edge weights of the multi-view communication network.
With the more attributions of ligands, HoloNet.tools.compute_ce_tensor() can set different w_best
for secreted ligands and plasma-membrane-binding ligands.
Then we filter the edges with low specificities.
elements_expr_df_dict = hn.tl.elements_expr_df_calculate(expressed_lr_df, complex_db, cofactor_db, adata)
ce_tensor = hn.tl.compute_ce_tensor(expressed_lr_df, w_best, elements_expr_df_dict, adata)
filtered_ce_tensor = hn.tl.filter_ce_tensor(ce_tensor, adata, expressed_lr_df, elements_expr_df_dict, w_best)
Note
This step will consume a lot of memory.
If you run out of memory, you can choose to only compute communication networks with fewer ligand-receptor pairs, and turn down the value of n_pairs parameter in HoloNet.tools.filter_ce_tensor()
Also, you can set copy=False in hn.tl.filter_ce_tensor or down-sample the dataset to reduced memory consumption.
When setting cut_porp as 2, we can run the whole tutorial on Macbook Pro (16G RAM, Apple M1 pro).
import random
cut_porp = 2
adata_subset = adata[random.sample(list(adata.obs_names), round(adata.shape[0] / cut_porp))]
elements_expr_df_dict = hn.tl.elements_expr_df_calculate(expressed_lr_df, complex_db, cofactor_db, adata_subset)
ce_tensor = hn.tl.compute_ce_tensor(expressed_lr_df, w_best, elements_expr_df_dict, adata_subset)
filtered_ce_tensor = hn.tl.filter_ce_tensor(ce_tensor, adata_subset, expressed_lr_df,
elements_expr_df_dict, w_best, copy=False)
Visualizing CEs#
Based on the multi-view CE network, we provide two visualization methods:
- CE hotspot plots for visualizing the centralities of spots. Provide two calculating methods:
Degree centrality: out-degree + in-degree, faithfully reflects the CE strength related to each spot.
Eigenvector centrality: reflects the core regions with active communication.
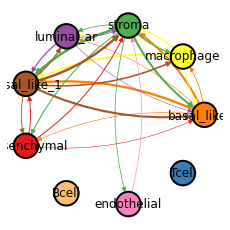
- Cell-type-level CE network:
CE strengths among cell-types
CEs hotspot plots#
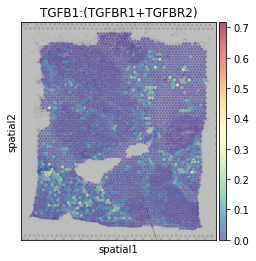
Degree centrality of each spot in the TGFB1:(TGFBR1+TGFBR2) CE network. Reflecting regions with active TGFB1:(TGFBR1+TGFBR2) communication.
hn.pl.ce_hotspot_plot(filtered_ce_tensor, adata,
lr_df=expressed_lr_df, plot_lr='TGFB1:(TGFBR1+TGFBR2)')

Hotspot plot based on eigenvector centrality. This plot better detects a clear center than the one based on degree centrality.
hn.pl.ce_hotspot_plot(filtered_ce_tensor, adata,
lr_df=expressed_lr_df, plot_lr='TGFB1:(TGFBR1+TGFBR2)',
centrality_measure='eigenvector')
Cell-type-level CE network#
Loading the cell-type percentage of each spot.
cell_type_mat, \
cell_type_names = hn.pr.get_continuous_cell_type_tensor(adata, continuous_cell_type_slot = 'predicted_cell_type',)
Plotting the cell-type-level CE network. The thickness of the edge represents the strength of TGFB1:(TGFBR1+TGFBR2) communication between the two cell types.
_ = hn.pl.ce_cell_type_network_plot(filtered_ce_tensor, cell_type_mat, cell_type_names,
lr_df=expressed_lr_df, plot_lr='TGFB1:(TGFBR1+TGFBR2)', edge_thres=0.2,
palette=hn.brca_default_color_celltype)
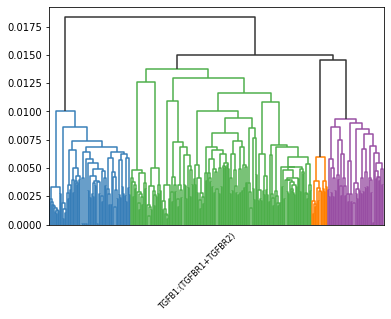
LR pair clustering#
Agglomerative Clustering the ligand-receptor pairs based on the centrality of each spot.
The cluster label of each ligand-receptor pair saved in clustered_expressed_LR_df['cluster'].
The number of clusters can be selected using n_clusters parameter in HoloNet.tools.cluster_lr_based_on_ce().
cell_cci_centrality = hn.tl.compute_ce_network_eigenvector_centrality(filtered_ce_tensor)
clustered_expressed_LR_df,_ = hn.tl.cluster_lr_based_on_ce(filtered_ce_tensor, adata, expressed_lr_df,
w_best=w_best, cell_cci_centrality=cell_cci_centrality)
Dendrogram for hierarchically clustering all ligand–receptor pairs.
hn.pl.lr_clustering_dendrogram(_, expressed_lr_df, ['TGFB1:(TGFBR1+TGFBR2)'],
dflt_col = '#333333',)
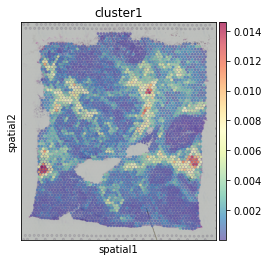
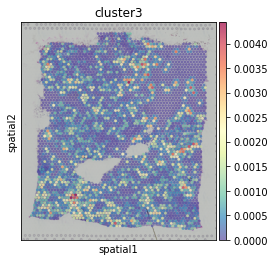
General CE hotspot of each ligand-receptor cluster (superimposing All CE hotspots of members in a cluster).
hn.pl.lr_cluster_ce_hotspot_plot(lr_df=clustered_expressed_LR_df,
cell_cci_centrality=cell_cci_centrality,
adata=adata)